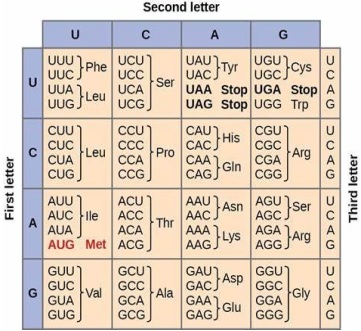
These genome sequences can be found in the NCBI database as of November 8th, 2023.
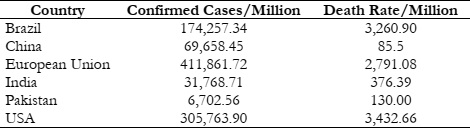
Table 1 provides an estimate of the number of confirmed cases and mortality rates for the
aforementioned countries. As the population of these countries differs, to compare death and
confirmed cases records, we have analyzed data per million records.
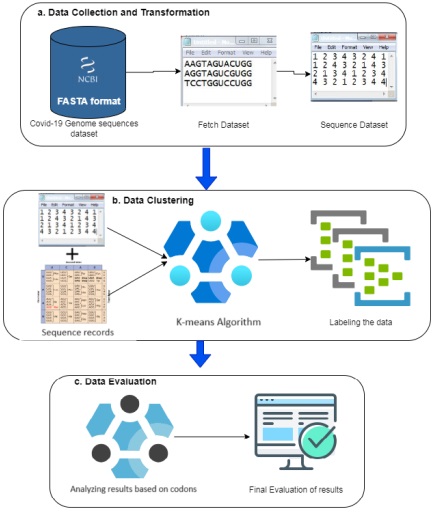
All the experiments were performed using the Bioinformatics library of Python. Figure
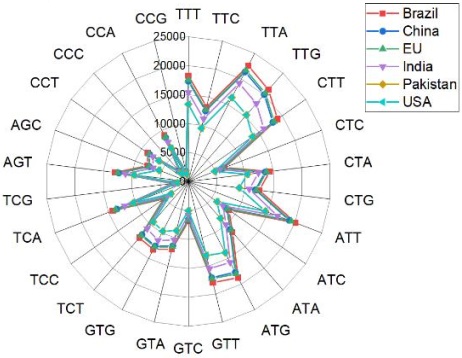
3 represents the distribution or frequency of specific amino acid codons across various
geographical locations or datasets before applying clustering algorithms. The counts in each cell
denote how many times a particular codon for a specific amino acid appeared in the
corresponding region. Based on the data presented in Figure 3, it can be observed that Leucine
(TTA) has the highest count of amino acids in raw data, occurring 22586 times in Brazil.
Similarly, Leucine (TTA) appears 21717 times in the EU and 21377 times in | (TTG) appears
21186 times in Brazil, while Cystine (TGT) appears 20568 times in Brazil, Leucine (TGT)
appears 20370 times in the EU, also, Isoleucine (ATT) appears 20092 times in Brazil. After
clustering, the count of frequent amino acids is limited compared to the huge count of codons
in the original data.
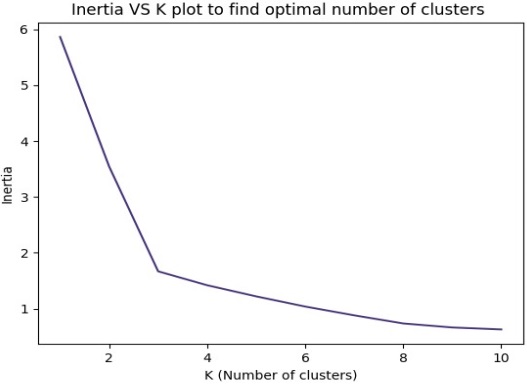
To apply the K-means algorithm [25], we need to determine the number of clusters,
denoted as "K". To achieve this, we can use the elbow method. This method involves plotting
a line graph between SSE (Sum of Squared Errors) and the number of clusters and identifying
the point where the graph changes direction as shown in Figure 4.. This point is known as the
"elbow point" and indicates the optimal number of clusters, as it is the point after which SSE or
inertia starts decreasing linearly. The elbow point in Figure 4 represents the point in the SSE /
Inertia plot where SSE or inertia starts decreasing, Here the elbow point is decreasing at point 3
so the value for k=3.
In this subsection, we have applied the TF-IDF method from the Python Bioinformatics
library [26]. It can be defined as calculating how relevant a word in a series or corpus is to a text.
The weight of a term that occurs in a document is simply proportional to the term frequency.
This tests the meaning of the text, which is very similar to TF, in the whole corpus
collection. The only difference is that in document d, TF is the frequency counter for a term
t, while df is the number of occurrences in the document set of the term t. In other words,
the number of papers in which the word is present is DF.
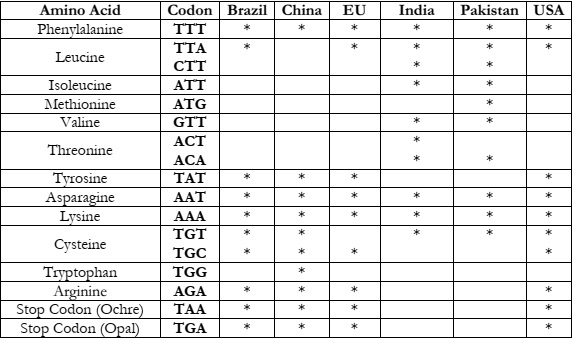
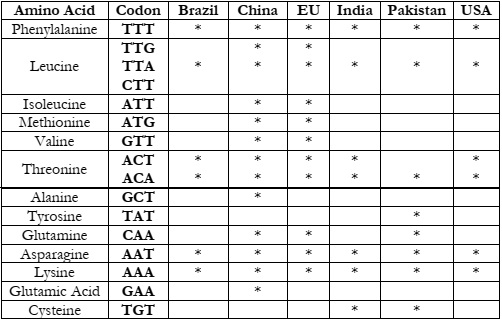
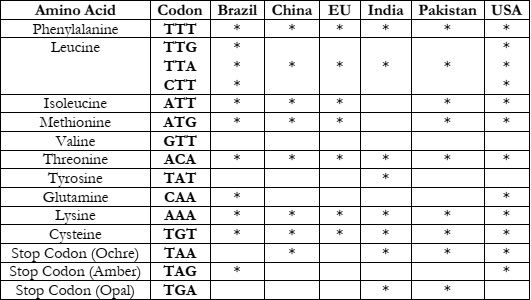
As the value of K is 3, we have obtained 3 clusters for each country. In Table 2-4, we
have listed the codons with each amino acid to study their occurrence after clustering. Table 2 displays the frequency of codons present in Cluster 1 for each country, and similarly, Table 3
and Table 4 represent Cluster 2 and Cluster 3, respectively.
It can be observed that Phenylalanine (TTT) and Lysine (AAA) are present in all
countries and each cluster so it’s a common amino acid present in all countries. In Table 3 (also
named Cluster 1), Asparagine (AAT) is present in all countries. Tryptophan (TTG) is only
present in China in Cluster 1 whereas Stop codon (Opal) and Arginine (AGA) are present in all
countries except India and Pakistan in Cluster 1. Cluster 1 also shows that in India and Pakistan
CTT, ATT, GTT and ACA are present but not in any other country. It is possible that these
codons are responsible for the similar effects observed in both countries.
In Table 4, Threonine (ACA) and Asparagine are present in all countries. TTG, ATG,
ATT, GTT, and CAA codons are mostly present in China and the European Union only. Stop
codons Opal and Ochre are present in all countries except for India and Pakistan. So the abovementioned codons if found in other countries may have similar effects for such pandemics that
are found similar in China and the EU.
In Table 5, Leucine, threonine, and Cysteine and Lycine amino acids are found in almost
all countries. But TTG, CTT, CAA, and TAG are mostly present in Brazil and the USA. As the
Brazil and USA are highly affected countries with higher mortality rates it can be observed that
these similar codons could be the reason for the similar effects of the COVID-19 pandemic.
Based on the previous results, it can be inferred that China and the EU have a lower
mortality rate due to their genome containing similar codons such as TTG, ATG, ATT, GTT,
and CAA from cluster 2, which are not commonly found in other countries. Similarly, countries
like Pakistan and India, with similar codons in their genome, such as CTT, ATT, GTT, and
ACA, have an average death rate and confirmed cases and can be considered medium-risk
countries for similar viruses. On the other hand, Brazil and the US have a higher mortality rate
and are highly affected countries with matching codons such as TTG, CTT, CAA, and TAG.
Therefore, countries with similar codons may have a high potential for such viruses. As in the
previous study [18], authors found some frequent amino acids in India, Pakistan, and China for
the Covid-19 genome sequence using the Clo Fast algorithm with varying min threshold, it
shows the availability of frequent codons in these 3 countries however our study tends to focus
on similar codons in different regions which gives better insight specifically for vaccination
purposes.
[1] D. Cucinotta and M. Vanelli, “WHO Declares COVID-19 a Pandemic,” Acta Biomed.,
vol. 91, no. 1, pp. 157–160, 2020, doi: 10.23750/ABM.V91I1.9397.
[2] A. N. Sajed and K. Amgain, “Coronavirus Disease (COVID-19) Outbreak and the
Strategy for Prevention,” Eur. J. Med. Sci. , vol. 2, no. 1, pp. 1–3, Mar. 2020, doi:
10.46405/EJMS.V2I1.38.
[3] F. Wu et al., “A new coronavirus associated with human respiratory disease in China,”
Nat. 2020 5797798, vol. 579, no. 7798, pp. 265–269, Feb. 2020, doi: 10.1038/s41586-
020-2008-3.
[4] “COVID-19 cases | WHO COVID-19 dashboard.” Accessed: Mar. 03, 2024. [Online].
Available: https://data.who.int/dashboards/covid19/cases?n=c
[5] S. Raskin, “Genetics of COVID-19,” J. Pediatr. (Rio. J)., vol. 97, no. 4, pp. 378–386, Jul.
2021, doi: 10.1016/J.JPED.2020.09.002.
[6] I. Fricke-Galindo and R. Falfán-Valencia, “Genetics Insight for COVID-19
Susceptibility and Severity: A Review,” Front. Immunol., vol. 12, p. 622176, Apr. 2021,
doi: 10.3389/FIMMU.2021.622176/BIBTEX.
[7] P. D. Cristea, “Conversion of nucleotides sequences into genomic signals,” J. Cell. Mol.
Med., vol. 6, no. 2, pp. 279–303, Apr. 2002, doi: 10.1111/J.1582-4934.2002.TB00196.X.
[8] F. Castro-Chavez, “Most Used Codons per Amino Acid and per Genome in the Code
of Man Compared to Other Organisms According to the Rotating Circular Genetic
Code,” Neuroquantology, vol. 9, no. 4, pp. 747–767, 2011, doi:
10.14704/NQ.2011.9.4.500.
[9] T. F. Clarke IV and P. L. Clark, “Rare Codons Cluster,” PLoS One, vol. 3, no. 10, p.
e3412, Oct. 2008, doi: 10.1371/JOURNAL.PONE.0003412.
[10] A. Baranova, H. Cao, S. Teng, K. P. Su, and F. Zhang, “Shared genetics and causal
associations between COVID-19 and multiple sclerosis,” J. Med. Virol., vol. 95, no. 1,
p. e28431, Jan. 2023, doi: 10.1002/JMV.28431.
[11] K. S. te Paske, C. van Tienen, D. Dunk, D. van Pelt, and P. W. Smit, “SARS-CoV-2
transmission among health care workers, an outbreak investigation using whole-genome
sequencing,” PLoS One, vol. 18, no. 3, p. e0283292, Mar. 2023, doi:
10.1371/JOURNAL.PONE.0283292.
[12] H. C. Maltezou et al., “SARS-CoV-2 Infection in Healthcare Personnel With High-risk
Occupational Exposure: Evaluation of 7-Day Exclusion From Work Policy,” Clin.
Infect. Dis., vol. 71, no. 12, pp. 3182–3187, Dec. 2020, doi: 10.1093/CID/CIAA888.
[13] J. Kim, S. Cheon, and I. Ahn, “NGS data vectorization, clustering, and finding key
codons in SARS-CoV-2 variations,” BMC Bioinformatics, vol. 23, no. 1, pp. 1–24, Dec.
2022, doi: 10.1186/S12859-022-04718-7/TABLES/4.
[14] B. Korber et al., “Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G
Increases Infectivity of the COVID-19 Virus,” Cell, vol. 182, no. 4, pp. 812-827.e19,
Aug. 2020, doi: 10.1016/J.CELL.2020.06.043.
[15] L. Zhang et al., “SARS-CoV-2 spike-protein D614G mutation increases virion spike
density and infectivity,” Nat. Commun. 2020 111, vol. 11, no. 1, pp. 1–9, Nov. 2020,
doi: 10.1038/s41467-020-19808-4.
[16] L. Guruprasad, “Human SARS CoV-2 spike protein mutations,” Proteins Struct. Funct.
Bioinforma., vol. 89, no. 5, pp. 569–576, May 2021, doi: 10.1002/PROT.26042.
[17] D. Mercatelli and F. M. Giorgi, “Geographic and Genomic Distribution of SARS-CoV-2 Mutations,” Front. Microbiol., vol. 11, p. 555497, Jul. 2020, doi:
10.3389/FMICB.2020.01800/BIBTEX.
[18] A. Umar, N. A. Mahoto, S. Bhatti, and S. Rathi, “Analysis of Covid-19 Genome
Sequences based on Geo-Locations,” Pakistan J. Eng. Technol., vol. 4, no. 4, pp. 41–45,
Dec. 2021, doi: 10.51846/VOL4ISS4PP41-45.
[19] M. R. Islam et al., “Genome-wide analysis of SARS-CoV-2 virus strains circulating
worldwide implicates heterogeneity,” Sci. Reports 2020 101, vol. 10, no. 1, pp. 1–9, Aug.
2020, doi: 10.1038/s41598-020-70812-6.
[20] N. Marascio et al., “Molecular Characterization and Cluster Analysis of SARS-CoV-2
Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One
Month,” Viruses 2023, Vol. 15, Page 802, vol. 15, no. 3, p. 802, Mar. 2023, doi:
10.3390/V15030802.
[21] D. A. Benson, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell, and D. L. Wheeler,
“GenBank,” Nucleic Acids Res., vol. 36, no. suppl_1, pp. D25–D30, Jan. 2008, doi:
10.1093/NAR/GKM929.
[22] J. A. Botía et al., “An additional k-means clustering step improves the biological features
of WGCNA gene co-expression networks,” BMC Syst. Biol., vol. 11, no. 1, pp. 1–16,
Apr. 2017, doi: 10.1186/S12918-017-0420-6/FIGURES/7.
[23] H. Z. Girgis, “MeShClust v3.0: high-quality clustering of DNA sequences using the
mean shift algorithm and alignment-free identity scores,” BMC Genomics, vol. 23, no.
1, pp. 1–16, Dec. 2022, doi: 10.1186/S12864-022-08619-0/FIGURES/3.
[24] A. Melnyk et al., “From Alpha to Zeta: Identifying variants and subtypes of SARS-CoV-
2 via clustering,” bioRxiv, p. 2021.08.26.457874, Aug. 2021, doi:
10.1101/2021.08.26.457874.
[25] N. Shi, X. Liu, and Y. Guan, “Research on k-means clustering algorithm: An improved
k-means clustering algorithm,” 3rd Int. Symp. Intell. Inf. Technol. Secur. Informatics,
IITSI 2010, pp. 63–67, 2010, doi: 10.1109/IITSI.2010.74.
[26] “Biopython · Biopython.” Accessed: Mar. 03, 2024. [Online]. Available:
https://biopython.org/